
A pdf version of this document created using latex can be downloaded by clicking here.
Abstract
Polymorphic malware detection is challenging due to the continual mutations miscreants introduce to successive instances of a particular virus. Such changes are akin to mutations in biological sequences. Recently, high-throughput methods for gene sequence classification have been developed by the bioinformatics and computational biology communities. In this paper, we argue that these methods can be usefully applied to malware detection. Unfortunately, gene classification tools are usually optimized for and restricted to an alphabet of four letters (nucleic acids). Consequently, we have selected the Strand gene sequence classifier, which offers a robust classification strategy that can easily accommodate unstructured data with any alphabet including source code or compiled machine code. To demonstrate Stand’s suitability for classifying malware, we execute it on approximately 500GB of malware data provided by the Kaggle Microsoft Malware Classification Challenge (BIG 2015) used for predicting 9 classes of polymorphic malware. Experiments show that, with minimal adaptation, the method achieves accuracy levels well above 95% requiring only a fraction of the training times used by the winning team’s method.
I. INTRODUCTION
The analogy between information security and biology has long been appreciated, since Cohen coined the term “computer virus” [4]. Modern malware frequently takes the form of a software program that is downloaded and executed by an unsuspecting Internet user. “Infection” can be achieved through compromising many thousands of websites en masse [15], social engineering, or by exploiting vulnerabilities on enduser systems. Regardless of how the infection occurs, cybercriminals have also undertaken considerable efforts to evade detection by antivirus software [16], [17].
Developers of such polymorphic malware attempt to avoid the detection of their malicious software by constantly changing the program’s appearance while keeping the functionality the same. This can be achieved by manipulating the code using multiple forms of obfuscation. Techniques include: encryption of malicious payloads, obfuscating variable names using character code shifts, equivalent code replacements, register reassignments, and removal of white space or code minification [3], [6], [22]. Individual instances of polymorphic malware can maintain the same general functionality while displaying many unique source code characteristics. For example, the computer worm Agobot or Gaobot was first identified around 2002 [27]. Over 580 variations of this malware were subsequently identified [2]. Today, each malware category can spawn many thousands of mutations, adding up to as much as one million new “signatures” per day [23]. In some cases, variations between malware instances occur simply to avoid detection. In others, new functionality emerges over time. Such changes require a robust form of malware classification which is less influenced by generational variation.
Gradual changes in polymorphic malware can be seen as mutations to the code. Thus, these changes are similar to mutation of biological sequences which occur over successive
generations.
Recently, significant advances have been made in gene sequence classification in terms of classification accuracy and processing speed. Originally, classification was based on expensive sequence alignment tools like BLAST [1] for comparing sample sequences to other sequences from known taxonomies. Many newer sequence classification tools claim
to be faster and/or more accurate. Examples are BLAT [13], the Ribosomal Database Project (RDP) naive Bayes classifier [26], UBLAST/USEARCH [7], Strand [5], Kraken [28], and CLARK [18].
Given the similarities between mutations in malware and in gene sequences, it stands to reason that the tools developed for gene sequence classification hold the potential to be applied to polymorphic malware detection. Consequently, in this paper we set out to apply one such classifier, called Strand (The Super Threaded Reference-Free Alignment-Free Nsequence Decoder) [5], to performing classification of polymorphic malware data. We selected Strand because, unlike the aforementioned gene sequence classifiers, it can process sequences of arbitrary alphabets. While BLAST has been adapted by researchers to process non-biological sequences [19], Strand can be used on general sequences “out of the box” and performs more efficiently than BLAST. We then use Strand to classify the malware dataset used in the Kaggle Microsoft Malware Classification Challenge (BIG 2015) [10]. We show how the application achieves comparable accuracy to the winning team’s malware classification techniques using only a fraction of the time required to generate the Strand training model.

II. BACKGROUND
Here we give a very short overview of word-based gene sequence classification which is typically done using word matching. Words are extracted from individual gene sequences and used for similarity estimations between two or more gene sequences [24]. Gene sequence words are sub-sequences of a given length. In addition to words they are often also referred to as k-mers or n-grams, where k and n represent the word length. The general concept of k-mers or words was originally defined as n-grams during 1948 in an information theoretic context [21] as a subsequence of n consecutive symbols. We will use the terms words or k-mers in this paper to refer to n-grams created from a gene sequence or other forms of unstructured input data. Over the past twenty years, numerous methods utilizing words for gene sequence comparison and classification have been presented [24].
Methods like BLAST [1] were developed for searching large sequence databases. Such methods search for seed words first and then expand matches. These so called alignment-free methods [24] are based on gene sequence word counts and have become increasingly popular since the computationally expensive sequence alignment method is avoided. The most common method for word extraction uses a sliding window of a fixed size. Once the word length k is defined, the sliding window moves from left to right across the gene sequence data producing each word by capturing k consecutive bases from the sequence.
The RDP classier [26] utilizes a fixed word length of only 8 bases to perform its taxonomy classification processing making the total possible number of unique words (i.e., features
for the classier) only 48 = 65; 536 words. Unfortunately, such a small feature space makes distinguishing between many sequence classes challenging as the probability for finding duplicate sequence words greatly increases when compared to the longer 30 base word length.
Rapid abundance estimation and sequence classification tools [18], [28] use longer words and derive a large speed advantage by utilizing an exact-match between the words extracted from sequence data to identify the similarity between two sequences. However, this approach comes at the cost of storing a very large number of sequence words to make accurate classifications when the value for k results in a very long word. For example, the extraction of k = 30 base words results in 4^30≈10^18 unique word possibilities within the training data feature space when an alphabet of 4 symbols (A;C; G; T) is considered. Other sequence classifiers avoid storing large volumes of words by reducing the value for k and the total possible feature space size for the training data
structure.
The issues with the need to store a large number of words becomes even more problematic when the size of the alphabet increases. This is clearly the case when we consider compiled code or source code. Strand addresses this problem by utilizing a form of lossy compression called Minhashing [8] which still supports sequence comparison, but with a much reduced memory footprint.
III. STRAND
Next, we give a very short overview of the Strand classification process (see [5] for more details).
Strand uses the map reduction aggregation process shown in Figure 1 to rapidly prepare and process input data in parallel during training or classification. Map reduction aggregation executes using shared memory during all stages within each Strand worker process. When multiple worker processes are used in a cluster, a single master process combines the outputs from each of the self-contained workers as they complete.
During stage 1 of map reduction aggregation, multiple threads extract words and associated classes from the gene sequence data in parallel. Simultaneously, a stage 2 combiner process minhashes each extracted word eventually creating a minhash signature for each input sequence provided. Finally, the unique minhash keys within each minhash signature are summarized by class during the reduce stage. During training, the reduce step adds minhash values into the training data structure, and during classification, minhash values are looked up within the training data structure and minhash intersections for each class are tabulated to determine one or more class similarity estimates.
A. Minhashing during Map Reduction Aggregation
Minhashing [8] is utilized within Strand to drastically reduce the amount of storage required for high capacity map reduction aggregation and classification function operations. Map reduction aggregation requires multiple pipeline stages when lossy compression via minhashing is deployed.
In Figure 1, Strand uses a map reduction aggregation pipeline including an additional combiner step to facilitate minhashing. This process also represents a more accurate method for Jaccard approximation than mere random selection of words. Minhashing is a form of lossy data compression used to remove a majority of the gene sequence words produced during stage one mapping by compressing all words into a much smaller minhash signature.
During stage one of the map reduction aggregation method shown in Figure 1, transitional sequence word outputs are placed into centralized, thread-safe storage areas accessible to minhash operation workers. In stage 2, a pre-determined number of distinct hashing functions are then used to hash each unique key produced during the stage one map operation one time each. As the transitional keys are repeatedly hashed, only one minimum hash value for each of the distinct hash functions are retained across all keys. When the process is completed only one minimum hash value for each of the distinct hash functions remains in a collection of minhash values which represent the unique characteristics of the learning or classification input data within a minhash signature.
To further enhance minhashing performance, only a single hash function can be used to generate a minhash signature. This eliminates the overhead of hashing words multiple times to support the family of multiple hashing functions traditionally used to create a minhash signature. In this scenario, all words are hashed by a single hashing function and n minimum hash values are selected to make up the minhash signature. These minimum values represent a random permutation of all words contained within the target sequence.

The minhash signature is further reduced by storing each minhash value in a partitioned collection of nested categorical key-value pairs. The training data structure illustrated in Figure 2 is designed in this manner. The training data structure’s nested key-value pairs are partitioned or sharded by each distinct hash function used. For example, when the minhashing process uses 100 distinct hash functions to create minhash signatures, the training data structure is divided into 100 partitions. All unique minhash keys created by hash function 0 are stored within partition 0 of the training data structure. Likewise, all unique minhash keys created by hash function 99 are stored in partition 99. However, when only a single hash function is used, no partitions are required.
The partitioned training data structure shown in Figure 2 includes minimum hash values which act as the key in the nested categorical key-value pair collection. Each minhash key contains as it value a collection of the classes which are associated with that key in the system. This collection of classes represents the nested categorical key-value pairs collection. Each nested categorical key-value pair contains a known class as its key and an optional frequency, weight, or any other numerical value which represents the importance of the association between a particular class and the minhash value key.
IV. CLASSIFICATION FUNCTION PROCESSING
Using a single training data structure, multiple classification scores are supported. Jaccard Similarity is calculated using the intersection divided by the union between the two sets. No frequency values are required for this similarity measure. For example, the Jaccard similarity between two sets S1 and S2 is defined as SJ (S1, S2), where:

Weighted Jaccard Similarity is also supported when the class frequency for unique minhash values are retained in the nested categorical key-value pair collection and taken into consideration [9]. The Weighted Jaccard similarity between two sets S1 and S2 is defined as SWJ (S1, S2), where:
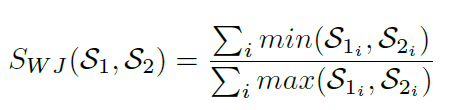
In Strand, Jaccard similarity is approximated by intersecting two sets of minhash signatures where longer signatures provide more accurate Jaccard similarity or distance approximations [20]. Class frequencies may be used to produce other Jaccard Index variations such as the Weighted Jaccard Similarity [9] shown above. However, large performance gains are achieved in Strand by using binary classification techniques where no nested categorical frequency values or log based calculations are required during classification function operations. In the binary minhash classification approach, minhash signature keys are simply intersected with the minhash keys of known classes to calculate the similarity between a query sequence and a known class.
After minhashing gene sequence words, we only have the sequence minhash signatures M1 = minhash(S1) and M2 = minhash(S2) representing the two sequences. Fortunately, minhashing [20] allows us to efficiently estimate the Jaccard index using only the minhash signatures:

where the intersection is taken hash-wise, i.e., how many minhash values agree between the two signatures.
Next, we discuss scoring the similarity between a sequence minhash signature and the category minhash signatures used for classification. Category signatures are not restricted to k values since they are created using the unique minhash values of all sequence minhash signatures belonging to the category. This is why we do not directly estimate the Jaccard index, but define a similarity measure based on the number of collisions between the minhash values in the sequence signature and the category signature.
Definition 1 (Minhash Category Collision): We define the Minhash Category Collision between a sequence S represented by the minhash signature M and a category signature C as:

where the intersection is calculated for each minhash hashing function separately.
We calculate MCC for each category and classify the sequence to the category resulting in the largest category collision count.
Many other more sophisticated approaches to score sequences are possible. These are left for future research.
V. APPLYING STRAND TO MALWARE CLASSIFICATION
The Kaggle Microsoft Malware Classification Challenge (Big 2015) [10] simulates the file input data processed by Microsoft’s real-time detection anti-malware products which are installed on over 160M computers and inspect over 700M computers each month [10]. The goal of the Microsoft Malware Classification Challenge is to group polymorphic malware at a high level into 9 different classes of malicious programs.
A. The Training and Classification Input Data
Microsoft provided almost a half terabyte of training and classification input data which included:
- Bytes Files: 10,868 training and 10,873 test .bytes files containing the raw hexadecimal representation of the file’s binary content with the executable headers removed.
- Asm Files: 10,868 training and 10,873 test .asm files containing a metadata manifest including data extracted by the IDA disassembler tool. This information includes things such as function calls, strings, assembly command sequences and more.
- Training Labels: Each training and test file name is a MD5 hash of the actual program. The training labels file contains each MD5 hash and the malware class which it maps to. No labels were provided for the test data input files.
B. Challenge Evaluation, Competitors, and Results
Kaggle challenge participants were evaluated using a multiclass logarithmic loss score. Each test file submission made required not only the predicted malware class, but the estimated probabilities for the file belonging to each of the 9 classes. Each submission record included the file hash and 9 additional comma-delimited fields containing values for the predicted probability that a given file belongs a particular class. The function logarithmic loss is defined as:

Where N is the number of test set files and M is the number of classes. The variable yij = 1 when file i is a member of class j and zero for all other classes. The predicted probability that observation i belongs to class j is given by the variable pij . The submitted probabilities are rescaled by pij = max (min (pij, 1-10^-15),10^-15) prior to scoring in order to avoid extremes in the log function [12].
There were 377 international teams competing in the contest with $16,000 in available prize money. The winning team achieved a logarithmic loss ratio score of 0.002833228 where a value of zero represents a perfect score. The winning team reported that their model produced an accuracy level greater than 99% during 10-fold cross-validation [11]. Their process was highly specialized and tailored specifically to the task and available data for detecting the 9 classes of malware presented in the challenge. Alternatively, we present here the results of a more general and performance oriented approach which should work well on many forms of input data where generational polymorphism occurs.
The winning team’s final submission used a highly complex ensemble of models [25] using a combination of features including: byte 4-gram instruction counts, function names and derived assembly features, assembly op-code n-grams, disassembled code segment counts, and disassembled code file pixel intensity. Generating these features required 500GB of disk space for the original training data and an additional 200GB for engineered features. While the final features used for the model required only 4GB, both feature engineering and generating the top performing model takes around 48 hours. Furthermore, it takes an additional 24 hours to generate the best model ensemble which produced the winning score [25]. In short, the winning submission takes 72 hours to produce.
C. Applying Strand to Microsoft Malware Classification Challenge
While Strand was originally designed to process FASTA format gene sequence files, only minor changes were required to accommodate for reading and processing the malware files as input. This is possible since unlike many other sequence classifiers and k-mer counters [14], [18], [28], Strand uses no special encoding of sequence data and supports any Unicode character within the sequences.
During gene sequence classification, the short reads of sequence data commonly generated by modern sequencers can be in either forward or reverse-complement order. As a result of this limitation, classification searches on sequence data must be made using each input sequence’s forward and reversecomplement effectively doubling the number of classification searches required. This particular feature of Strand is gene sequence specific and was irrelevant for malware classification. After turning off the reverse-complement search and modifying the sequence file parsing routine, Strand was able train and classify against malware data with no other changes.
D. Developing Malware Features for Strand
All of the malware feature engineering required to convert malware program data into Strand sequences, fits into just a few lines of code. First, the disassembled code files (.asm files) were not used to produce the score and accuracy results presented later in Tables I and II. The disassembled code files were eliminated since they did not increase the accuracy score when using them in combination with the binary content of the malware (.bytes files). In the future, a two model ensemble could be created which classifies each malware file in Strand based on combining the individual scores from dual .bytes and .asm model predictions.

Figure 3 illustrates the typical content encountered within the .bytes hex data files provided by Microsoft. The first 8 characters of each line contain a line number, and the last line shows how some hex content is unavailable and displayed as “??”. Both the line numbers and “??” symbols are removed during Strand processing.

When reading each .bytes file, Strand uses the code shown in Figure 4 to convert the .bytes malware files into a Strand sequence. During processing each carriage return, space, and
“?” character are removed. This produces a single string or Strand sequence containing all hex content read from the malware file. Once the malware hex data is cleaned, sequence words of length k or k-mers are generated by Strand as previously described.
E. Malware Classification Results Using Strand
While we did not produce a winning logarithmic loss score for the Kaggle Microsoft Malware Classification Challenge (BIG 2015) [10], we were able achieve a score of 0.452784 when using a 32-bit minhashing configuration and a score of 0.222864 when using a 64-bit minhashing configuration with Strand. We used a word length of 10 characters and 2400 minhash values within the Strand minhash signature to achieve this result. The primary benefit of using Strand was achieving an acceptable degree of accuracy within a short period of time. Training and classification times were both under 7 hours for processing 224GB of training data and 189GB of test data.
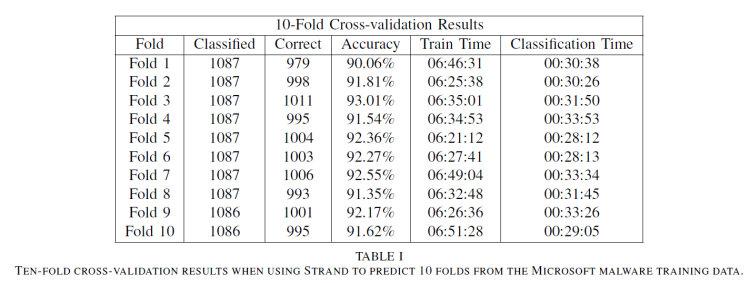
Table I shows ten-fold cross-validation results for the Malware Classification Challenge training data. Strand averaged 91.88% accuracy across the ten folds predicted using only 32-bit hashing functions. When using 64-bit hashing functions, we were able to drastically reduce the logarithmic loss score produced from 0.452784 to 0.222864. Table II shows tenfold cross-validation results for the version of Strand using 64-bit hash codes. Strand averaged 97.41% accuracy across the ten folds. While memory consumption increased slightly, there was no large degradation in training or classification performance.

The training times in Tables I and II represent the time required to train on 90% of the 10,868 Malware Classification Challenge training data records (9,782 training records). The
classification times in both tables reflect the time required to classify the number of records reflected in the “Classified” column which represent 10% of the training data for each fold. The 32-bit and 64-bit versions of Strand required 5.482 and 5.483 total hours respectively to classify all of the 10,868 training records during ten-fold cross validation.
The 64-bit model takes up approximately 5GB in memory and 436MB compressed on disk while the 32-bit version takes up approximately 3GB in memory and 255MB compressed on disk. Due to the small size of the model, multiple copies can be loaded into memory for multiple worker processes to take advantage of process level parallelism when classifying large volumes of data. For example, fifteen classification workers were used to process the test files provided by Microsoft.
VI. CONCLUSION
In this paper we have demonstrated how modern gene sequence classification tools can be applied to large-scale malware detection. In this first study, we have shown how the gene sequence classifier Strand can be used to predict multiple classes of polymorphic malware using data provided by the Kaggle Microsoft Malware Classification Challenge (Big 2015). While the approach, using only minimal adaptation, did not best the accuracy scores achieved by the highly tailored approach that won the competition, we did achieve classification accuracy levels exceeding 95% while making predictions in less than 10% of the training times required by the winning team.
From the success of this demonstration, we conclude that gene sequence classifiers in general, and Strand in particular, hold great promise in their application to security datasets. In addition to polymorphic malware, we anticipate that these classifiers can be used anywhere data sequences are used, such as in network traces of attacks.
REFERENCES
[1] S. F. Altschul, W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. Basic
local alignment search tool. Journal of Molecular Biology, 215(3):403–
410, 1990.
[2] M. Bailey, J. Oberheide, J. Andersen, Z. M. Mao, F. Jahanian, and
J. Nazario. Automated classification and analysis of internet malware. In
Recent advances in intrusion detection, pages 178–197. Springer, 2007.
[3] M. Christodorescu, S. Jha, S. Seshia, D. Song, R. E. Bryant, et al.
Semantics-aware malware detection. In Security and Privacy, 2005 IEEE
Symposium on, pages 32–46. IEEE, 2005.
[4] F. Cohen. Computer viruses. Comput. Secur., 6(1):22–35, Feb. 1987.
[5] J. Drew and M. Hahsler. Strand: fast sequence comparison using
mapreduce and locality sensitive hashing. In Proceedings of the 5th
ACM Conference on Bioinformatics, Computational Biology, and Health
Informatics, pages 506–513. ACM, 2014.
[6] J. M. Drew. Mass compromise of iis shared web hosting for blackhat
seo: A case study, 2014. https://blog.jakemdrew.com/2015/03/10/
mass-compromise-of-iis-shared-web-hosting-for-blackhat-seo-a-case-study/
;AccessedOn:01/06/2016.
[7] R. C. Edgar. Search and clustering orders of magnitude faster than blast.
Bioinformatics, 26(19):2460–2461, 2010.
[8] A. Gionis, P. Indyk, R. Motwani, et al. Similarity search in high
dimensions via hashing. In VLDB, volume 99, pages 518–529, 1999.
[9] S. Ioffe. Improved consistent sampling, weighted minhash and l1
sketching. In Data Mining (ICDM), 2010 IEEE 10th International
Conference on, pages 246–255. IEEE, 2010.
[10] Kaggle. Microsoft malware classification challenge (big 2015), 2015.
https://www.kaggle.com/c/malware-classification;Accessed:2015-11-04.
[11] Kaggle. Microsoft malware winners’ interview: 1st place,
“no to overfitting”, 2015. http://blog.kaggle.com/2015/05/26/
microsoft-malware-winners-interview-1st-place-no-to-overfitting/
;Accessed:2015-11-02.
[12] Kaggle. Evaluation, 2016. https://www.kaggle.com/c/
malware-classification/details/evaluation;AccessedOn:01/14/2016.
[13] W. J. Kent. Blat-the blast-like alignment tool. Genome research,
12(4):656–664, 2002.
[14] G. Marcais and C. Kingsford. A fast, lock-free approach for efficient
parallel counting of occurrences of k-mers. Bioinformatics, 27(6):764–
770, 2011.
[15] N. P. P. Mavrommatis and M. A. R. F. Monrose. All your iframes point
to us. In USENIX Security Symposium, pages 1–16, 2008.
[16] McAfee. For consumers, 2014. https://www.mcafee.com/consumer/
en-us/store/m0/index.html;AccessedOn:01/06/2016.
[17] Norton. Norton anti, 2014. http://us.norton.com/;AccessedOn:01/06/
2016.
[18] R. Ounit, S. Wanamaker, T. J. Close, and S. Lonardi. Clark: fast and
accurate classification of metagenomic and genomic sequences using
discriminative k-mers. BMC genomics, 16(1):236, 2015.
[19] E. Peterson, D. Curtis, A. Phillips, J. Teuton, and C. Oehmen. A generalized
bio-inspired method for discovering sequence-based signatures.
In Intelligence and Security Informatics (ISI), 2013 IEEE International
Conference on, pages 330–332, June 2013.
[20] A. Rajaraman and J. Ullman. Mining of Massive Datasets. Mining of
Massive Datasets. Cambridge University Press, 2012.
[21] C. E. Shannon. A mathematical theory of communication. ACM
SIGMOBILE Mobile Computing and Communications Review, 5(1):3–
55, 2001.
[22] P. Ször and P. Ferrie. Hunting for metamorphic. In Virus Bulletin
Conference, 2001.
[23] V. Total. File statistics during last 7 days. https://www.virustotal.com/
en/statistics/. Last retrieved 2015-01-15.
[24] S. Vinga and J. Almeida. Alignment-free sequence comparison — A
review. Bioinformatics, 19(4):513–523, 2003.
[25] L. Wang. Microsoft malware classification challenge
(big 2015) first place team: Say no to overfitting, 2015.
https://github.com/xiaozhouwang/kaggle_Microsoft_Malware/blob/
master/Saynotooverfitting.pdf;Accessed:2015-11-02.
[26] Q. Wang, G. M. Garrity, J. M. Tiedje, and J. R. Cole. Naive bayesian
classifier for rapid assignment of RNA sequences into the new bacterial
taxonomy. Applied and Environmental Microbiology, 73(16):5261–5267,
2007.
[27] Wikipedia. Agobot, 2014. https://en.wikipedia.org/wiki/Agobot;
AccessedOn:01/06/2016.
[28] D. E. Wood and S. L. Salzberg. Kraken: ultrafast metagenomic sequence
classification using exact alignments. Genome Biol, 15(3):R46, 2014.
